In the past 20 years, RGS proteins have emerged as novel drug targets in numerous disease states, such as hypertension, cancer and Parkinson’s disease among others. This is not surprising given that 40-60% of currently FDA approved drugs target GPCR signaling. RGS protein levels and activity are tightly regulated in space and time by post-translational modifications, as well as transcriptional and epigenetic regulation and proteasomal degradation. Furtermore, several RGS proteins are down-regulated in disease states, leading to dysregulated GPCR signaling. Work in our lab aims to identify mechanisms by which RGS protein levels and activity are controlled and developing strategies to target these in drug discovery efforts. Enhancing RGS protein activity leads to suppression of GPCR signaling in a manner that is dependent on receptor activity. Thus, modulating RGS proteins would not create signals in the cells, rather modulating and fine-tuning signals occurring through the receptor.
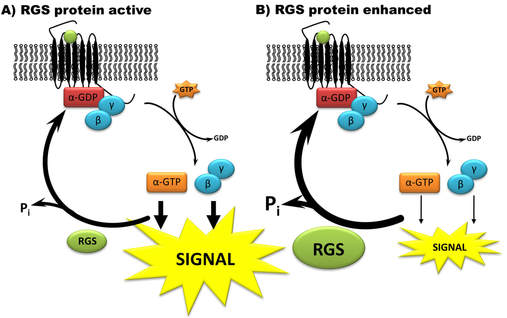
RGS proteins all share a common RGS domain that directly interacts with active, GTP-bound Gα subunits of heterotrimeric G proteins. RGS proteins stabilize the transition state for GTP hydrolysis on Gα and thus induce a conformational change in the Gα subunit that accelerates GTP hydrolysis, thereby effectively turning off signaling cascades mediated by GPCRs. Many efforts have been made to develop inhibitors of the Gα-RGS interaction. Our lab instead focuses on finding strategies to enhance RGS protein function through modulation of the mechanisms that control activity and/or expression.
RGS proteins all share a common RGS domain that directly interacts with active, GTP-bound Gα subunits of heterotrimeric G proteins. RGS proteins stabilize the transition state for GTP hydrolysis on Gα and thus induce a conformational change in the Gα subunit that accelerates GTP hydrolysis, thereby effectively turning off signaling cascades mediated by GPCRs. Many efforts have been made to develop inhibitors of the Gα-RGS interaction. Our lab instead focuses on finding strategies to enhance RGS protein function through modulation of the mechanisms that control activity and/or expression.
Mechanisms of RGS2 protein degradation
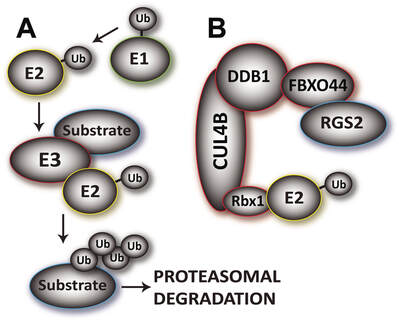
We have a long-time interest in understanding the mechanisms involved in regulating RGS2 function with a view to develop therapies for diseases involving RGS2 dysfunction. A key mechanism controlling RGS2 protein levels is through rapid degradation by the ubiquitin-proteasomal system (UPS). The UPS plays an essential role in cell functions and dysregulation often manifests in pathophysiology. Through a series of enzymes (E1, E2 and E3’s) a chain of ubiquitin molecules is built onto the target protein, signaling the proteasome to degrade it. The UPS is receiving immense interest in drug discovery, with the development of proteasome inhibitors for the treatment of several cancers. Not surprisingly, completely inhibiting such an important system leads to severe side effects. Over 600 E3 ligases are responsible for recognizing and recruiting proteins to be degraded and confer specificity within the UPS. We identified an E3 ligase targeting RGS2 for degradation. The RGS2-recruiting component of this complex is F-box protein 44 (FBXO44). Inhibiting the RGS2-FBXO44 interaction is a viable and selective approach to stabilize RGS2 protein. We are currently pursuing several avenues of investigation in this area. This includes biochemical and structural characterization of the RGS2-E3 ligase interactions, as well as assay development and screening for small molecule inhibitors of the RGS2-FBXO44 interaction.
This project is sponsored by NIH grant R01GM143493, and has previously received funding from the American Heart Association, The Swedish Heart- and Lung foundation and the Ralph W. and Grace M. Showalter Research Trust.
This project is sponsored by NIH grant R01GM143493, and has previously received funding from the American Heart Association, The Swedish Heart- and Lung foundation and the Ralph W. and Grace M. Showalter Research Trust.
Drug discovery for RGS10 protein enhancement
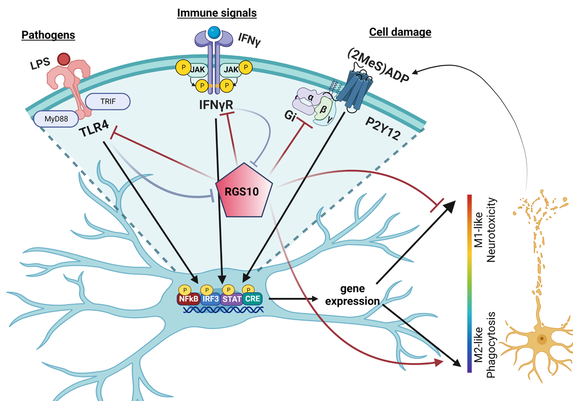
Neuroinflammation, mediated by microglia, is a major component of several neurological diseases, including Parkinson’s disease, Alzheimer’s disease, ALS and chronic pain, all lacking effective, disease-modifying therapies. Regulator of G Protein Signaling 10 (RGS10) serves a key protective role in neuroinflammation, in that it suppresses inflammatory signaling in microglia and reduces neurotoxicity resulting from chronic neuroinflammation. Targeting dysregulated inflammatory signaling in microglia is a promising therapeutic strategy; however, strategies regulating individual inflammatory signaling pathways have lacked clinical efficacy. The microglial protein Regulator of G protein Signaling 10 (RGS10) inhibits pro-inflammatory signaling downstream of TLR4 and TNFalpha receptors, and protects against inflammation-induced neurotoxicity. RGS10 expression is strongly suppressed through epigenetic mechanisms following microglial activation and this silencing amplifies microglial signaling in a feed-forward mechanism that contributes to dysregulation of inflammatory signaling and contributes to chronic neuroinflammation. Blocking the suppression of RGS10 expression in activated microglia will diminish multiple inflammatory pathways and limit neuroinflammation. We are developing a non-biased high-throughput screening (HTS) assay to identify and validate small molecule regulators of RGS10 expression in resting and activated microglia. We are also using bioinformatic approaches to determine the mechanisms regulating RGS10 in microglia and, in turn, how RGS10 regulates microglial function.
This project was previously funded by the National Institutes of Health (NIA 1R21AG064416-01)
This project was previously funded by the National Institutes of Health (NIA 1R21AG064416-01)